
Diagnosis
Suspicion of amyloidosis
Suspicion of amyloidosis can arise in many ways. For example, if someone belongs to a family with hereditary amyloidosis, minor symptoms can already lead to a strong suspicion of the presence of amyloidosis. If someone is known to have a long-term inflammatory disease such as rheumatoid arthritis, it is good to monitor the possible development of AA amyloidosis by regularly checking the urine for the presence of protein and monitoring kidney function. Unexplained enlargement of the tongue, liver or spleen, neuropathy of both legs, severe loss of protein via the kidneys or kidney failure, severe diarrhea, or heart failure with strongly thickened heart walls can also sometimes lead to suspected amyloidosis. This is even more true with combinations of the above-mentioned unexplained symptoms.
Suspicion alone is insufficient, the presence of amyloid should be unambiguously determined in a biopsy.
Often, however, the result of such a biopsy is the first evidence amyloidosis. Due to the relative rarity of amyloidosis, the widely different ways in which the disease can present itself and the insidious and very gradual development of disease symptoms, in many cases the disease is only discovered at a late stage, in which serious damage has already been done to one or more organs.
Further assessment in brief
After the discovery of amyloid in a tissue sample, further evaluation should take place to identify the type of amyloidosis, and map its severity and extent. It is important to determine whether the amyloid accumulation is localised or systemic, and which organs and tissues may have been damaged by it. This provides the practitioner with a picture of the severity and extent of the amyloidosis and allows a rough estimate of the life expectancy to be made. From there, it can be considered whether treatment is possible and, if so, what the options are.
Biopsy
In order to determine the presence of amyloid, it is necessary to identify it in tissue samples. Amyloid can be detected by a positive staining using Congo Red dye, and the associated apple-green birefringence in polarised light. An abdominal fat biopsy is the most elegant and least invasive method for this purpose, with a >90% chance of detection of most forms of systemic amyloidosis, if the biopsy and the processing of it are carried out correctly. A good alternative is to perform a rectal biopsy. If one of these methods gives a negative result, but a strong suspicion of amyloidosis remains, then it is worthwhile to carry out the other method (or take a bone marrow biopsy, a somewhat less reliable method of detection). However, if all these biopsies are negative and the suspicion of amyloid nevertheless persists, then a biopsy of the suspected organ or tissue is appropriate.
Below is an example of the Congo Red coloring of a suction (aspiration) biopsy of fat tissue. This sample can be taken at an outpatient clinic under local anesthetic, just beneath the skin of the stomach close to the navel (bellybutton). Download the instruction video (approx. 15 MB) here and see the Fat aspiration procedure 2019 (pdf file) for more details.
 |
 |
Fat aspiration – normal light | Fat aspiration – polarised light |
As can be seen in the above images, the amyloid is visible as red-colored deposits between the normal, blue-colored structure of the fat tissue. When viewed under polarised light, the red color changes to green or yellow-green. The green birefringence of material dyed with Congo Red under polarised light is characteristic of amyloid.
In the laboratory, the presence and quantity of the precursor proteins can then be examined. It is also possible to look under a microscope in tissue pieces of the affected organ on a specimen slide with specific antibodies (directed against the precursor proteins) in order to establish the presence of amyloid irrefutably and to investigate which type of amyloid is involved.
Typing
Clinical assessment of the type of amyloid
After confirmation of the presence of amyloid in tissue, the amyloid should be classified. In many cases, a good assessment of the type of amyloid can be made based on medical history, symptoms and clinical picture. A patient with long-standing rheumatoid arthritis and nephrotic syndrome almost certainly has the AA type. Someone with polyneuropathy who belongs to a family with hereditary amyloidosis is likely to have the ATTR type. And for a patient with the distinctive swelling of both shoulders (“shoulder pads”) and a significantly enlarged tongue, the AL type is most likely. Blood, urine and bone marrow tests can also help to predict the type of amyloidosis. In thre case of AL amyloidosis, proteins produced by malignant plasma cells (free light chains) can almost always be detected in the blood and / or urine and malignant plasma cells can be detected in the bone marrow. With AA amyloidosis, elevated acute phase proteins (such as CRP and the precursor protein SAA) can be found in the blood. This is indicative of, but certainly not proof of AA amyloidosis, as these inflammatory proteins may also be present in the blood at high levels in many other circumstances.
Even when a certain type of amyloid is clinically plausible, confirmation of the amyloid type is necessary. The treatment, prognosis and consequences of the different types of systemic amyloidosis vary considerably.
Typing using biopsy
Immunohistological examination of the biopsy is a technique in which the type of amyloid can be further characterized via specifically targeted antibodies. In the case of AA amyloidosis, this test is sufficient for typing, provided that reliable antibodies are used for this. However, with AL and ATTR amyloidosis, this method fails in some cases because the antibodies used are less reliable and the recognizability of the precursor protein within the amyloid deposits is reduced.
Below are examples of a kidney biopsy of an AA amyloidosis patient where the biopsy is stained with Congo Red (where the characteristic effect of the polarization filter can be seen) and on the right the Immunohistochemical staining with the reliable antibody directed against Serum amyloid A (SAA; clone Reu86.2).
 |
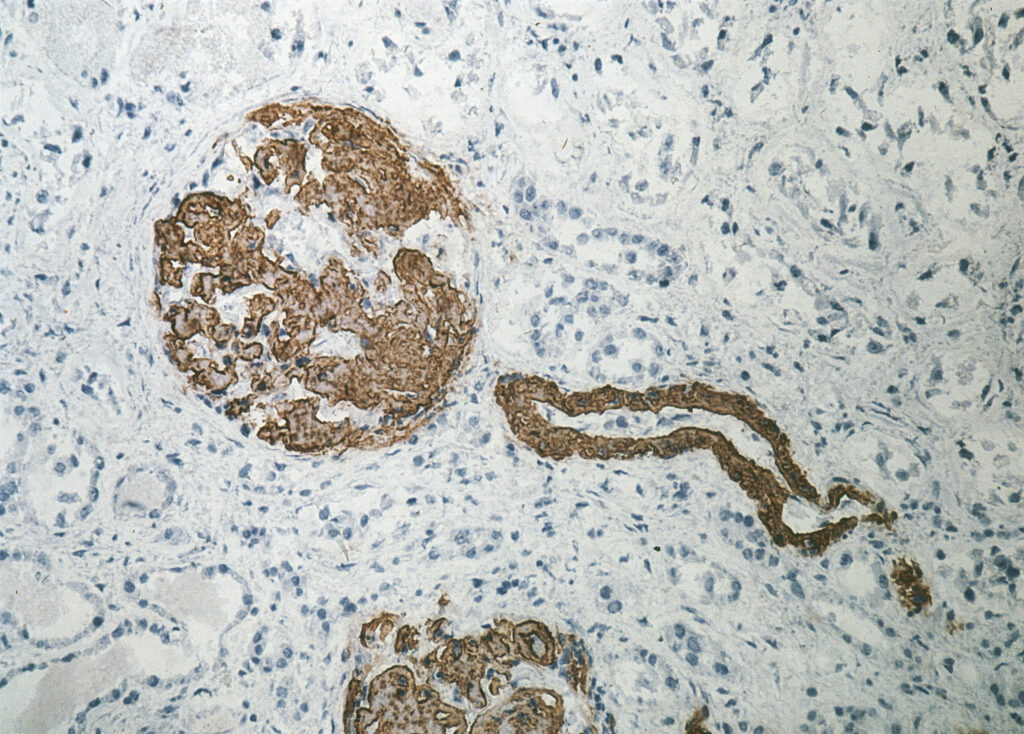 |
Kidney – Congo Red staining | Kidney – Immunohistochemical staining with anti-SAA |
In Groningen we are able to make the amyloid available by means of certain solvents from the fat biopsy and to type the amyloid using four laboratory tests, so-called ELISAs. This method is suitable for distinguishing the most common types of amyloidosis (AA, AL and ATTR), but it only works if there is sufficient amyloid in the fat biopsy. If this method fails to type the amyloid, other biochemical techniques (such as mass spectrometry) can be condidered.
Genetic testing
If there is evidence of ATTR amyloidosis, DNA testing will distinguish between the hereditary and the wild-type form. In addition to hereditary ATTR amyloidosis, there are several other, rarer types of hereditary amyloidosis (see table).
The DNA testing is done at the Department of Clinical Genetics. Most forms of hereditary amyloidosis have a autosomaal dominant inheritance pattern. Autosomal means that the predisposition is not on the sex chromosomes, but on one of the other chromosome pairs. Dominant means that the predisposition in only one of the two chromosomes of a chromosome pair is sufficient to express the disease. The gene on the other chromosome therefore has no predisposition. This means that children, both sons and daughters, of a parent with a predisposition to hereditary amyloidosis, each have a 50% chance of inheriting this predisposition. However, having the hereditary predisposition does not always mean that hereditary amyloidosis is also manifested.
When hereditary amyloidosis is diagnosed, adult family members are eligible for DNA testing. Patients with hereditary amyloidosis receive an information letter that they can give to their relatives. Family members who wish to have DNA testing for hereditary amyloidosis and / or would like more information about this can be referred to the Department of Clinical Genetics by their general practitioner. If a DNA test shows that someone is a carrier of the predisposition for hereditary amyloidosis, a referral is made to the expertise center for screening for possible symptoms.
| Abbreviation | Protein (precursor) |
Affected organs |
| ApoAI | Apolipoprotein A-I | Heart, liver, kidneys, nerves, larynx |
| ApoAII | Apolipoprotein A-II | Kidneys, heart |
| ApoCII | Apolipoprotein C-II | Kidneys |
| ApoCIII | Apolipoprotein C-III | Kidneys |
| Beta2M | Beta-2 microglobulin | Gastrointestinal tract, autonomic nervous system, salivary gland, lacrimal glands (tear ducts) |
| CST3 | Cystatin | Brain |
| FGA | Fibrinogen A α-chain | Kidneys |
| GSN | Gelsolin | Nervous system, skin and eyes |
| IL31RA | Interleukin-31 receptor A | Skin |
| LYS | Lysozyme | Kidneys, liver, heart, spleen, gastrointestinal tract and salivary gland |
| OSMR | Oncostatin M receptor | Skin |
| TTR | Transthyretin | Nervous system, heart, eyes, meninges |
In brief
- If amyloidosis is suspected, the amyloid must be proven to be present in a biopsy
- The type of amyloid must also be determined. Various laboratory techniques are available for this
- Genetic testing is performed to assess whether the form of amyloidosis is hereditary
