
Diagnosis
Suspicion of amyloidosis
The detection and determination of the presence of amyloid is preceded by the emergence of a clinical suspicion of amyloidosis, see Table. Clinical suspicion of amyloidosis can arise in many ways.
| Category | Clinical situation | Symptoms and signs | Type of amyloïd | Preferred biopsy |
| I | Unexpected and unexplained or already known with a plasma cell dyscrasia |
Proteinuria, loss of renal function, diarrhea, organomegaly (liver, spleen, or tongue), cardiac failure, biventricular thickened cardiac walls, cardiac rhythm or conduction problems, orthostatic hypotension, peripheral axonal polyneuropathy, autonomic neuropathy, carpal tunnel syndrome, severe weight loss, or malabsorption | AL | Primary: abdominal fat #; Secondary: Involved tissue& | II | Known with longstanding inflammation | Proteinuria, loss of renal function, cardiac failure, diarrhea, hepatomegaly, or splenomegaly | AA | Primary: abdominal fat #; Secondary: Involved tissue& |
| III | Member of a family known with hereditary amyloidosis | Symptoms or signs related to that particular type of amyloidosis. Some examples: Cardiomyopathy in ATTRv, Nephropathy in AFib, and Cornea lattice dystrophy in AGel | Erfelijke typen, zoals ATTRv, AApoAI, AFib, AGel, en andere* | Primary: abdominal fat #; Secondary: Involved tissue& | IV | An elderly (often male) patient without known cardiovascular disease | Cardiac failure with a history of carpal tunnel syndrome, atrial fibrillation, aortic stenosis, or spinal stenosis | ATTRwt | Endomyocardial tissue or bone scintigraphy$ |
| V | A tumor localized in specific sites of the body | Larynx or bronchial tree, urogenital tract, eyelid, gastrointestinal tract, pharynx | local AL | Involved tissue | VI | Eosinophilic material in a biopsy: amyloid suspected | Not beforehand recognized as such by the applicant physician. Awareness of the pathologist | All types | NA |
#The general opinion, although some centers prefer lip (accessory salivary glands) or rectum.
&Rectum, gastrointestinal (unspecified) or lip are also mentioned as alternative or step in-between.
*See: Benson M. et al. Amyloid nomenclature 2020. Amyloid 2020; 27:217-22.
$When AL amyloidosis has been excluded by negative IFE of serum and urine and normal immunoglobulin free light chains in serum.
NA, not applicable.
The first category consists of people who have developed new symptoms or other symptoms that do not fit their medical history. An unexplained enlargement of the tongue, liver or spleen, a disturbed functioning of nerves (neuropathy) in both legs, severe loss of protein via the kidneys or kidney failure, severe diarrhea, or heart failure with heavily thickened heart walls can sometimes lead to suspected amyloidosis. Suspicion increases with more than one symptom.
A second category consists of patients with a long-standing inflammatory picture, such as rheumatoid arthritis. Then it is good to be aware of the possible development of AA amyloidosis by regularly checking the urine for the presence of protein and keeping a close eye on kidney function.
A third category consists of members of a family in which hereditary amyloidosis occurs and who have developed symptoms that are consistent with that type of amyloidosis.
A fourth category consists of elderly (mostly male) patients with heart failure without a cardiovascular history, but with a different characteristic story.
A fifth category consists of patients with one or more swellings that are located in specific areas of the body.
The last, but not the least frequent, category consists of people whose amyloidosis is detected by an alert pathologist. After finding suspicious eosinophilic material in a tissue biopsy, he decides to do a staining with Congo red, resulting in amyloid being found.
Suspicion alone is not enough, the presence of amyloid must be unambiguously established in a piece of tissue, a so-called biopsy.
Often the result of such a biopsy is the first finding that there is amyloidosis. Due to the relative rarity of amyloidosis, the very different ways in which the disease can present and the insidious and very gradual onset of disease symptoms, the disease is in many cases only discovered at a late stage, when serious damage has already been done to one or more organs.
Further evaluation in brief
After finding amyloid in a piece of tissue, further investigation should be carried out by typing and mapping the severity and extent of the amyloidosis. It is important whether the amyloid is stacked locally or systemically and which organs and tissues are involved and possibly damaged in this stacking. This provides a picture of the severity and extent of the amyloidosis and an overall estimate of the prognosis can be made. Based on this situation, it should be examined whether there are possibilities for treatment and, if so, which options these are.
Biopsy
To determine the presence of amyloid, it is required to demonstrate this in tissue sections. This is possible by a positive coloring with Congo Red, with the typical apple-green birefringence in polarized light. For this purpose, an abdominal fat biopsy is the most elegant and least burdensome method, with a >90% chance of detection of most forms of systemic amyloidosis, if the biopsy and the processing of it are properly performed. A good alternative is to perform a rectal biopsy. If one of these methods gives a negative result, but a strong suspicion of amyloidosis remains, then it is worthwhile to also carry out the other one (or try the somewhat less reliable bone marrow biopsy). However, if all these biopsies are negative yet the suspicion of amyloid nevertheless persists, then a biopsy of the suspected organ or tissue is appropriate.
Below are examples of the Congo Red staining of aspirated adipose tissue. This tissue can be taken on an outpatient basis under local anesthetic, slightly next to the navel and just below the abdominal skin. Download the instruction video here (about 15 MB) and view the Fat aspiration procedure 2019 (pdf file) for more details.
 |
 |
Fat aspirate – normal light | Fat aspirate – poralised light |
As can be seen in the figures above, the amyloid in the adipose tissue is recognizable as red-colored deposits between the normal, slightly blue-colored architecture of adipose tissue. When viewed in polarized light, the red color changes to green or green-yellow. This green birefringence of Congo-red colored material in polarized light is characteristic of amyloid.
The amyloid can then be extracted from the biopsy in the laboratory and the presence and quantity of precursor proteins can be examined. Tissue sections of the affected organ can also be examined under the microscope with specific antibodies (directed against the precursor proteins) to irrefutably determine the presence of amyloid and to investigate the type of amyloid involved.
Typing
Clinical assessment of the type of amyloid
After confirmation of the presence of amyloid in tissue, typing should follow. In many cases, a good estimate can already be made of the type of amyloid on the basis of medical history, complaints and clinical picture. A patient with long-standing rheumatoid arthritis and a nephrotic syndrome almost certainly has the AA type. Someone with polyneuropathy who belongs to a family with hereditary amyloid is likely to have the ATTR type. And in a patient with the characteristic “shoulder pads” and a significantly enlarged tongue, the AL type is most likely. Blood, urine and bone marrow tests can also help to assess what type of amyloidosis is involved. AL-amyloidosis can often be determined by free light chains in the blood and/or urine. Clonal plasma cells can be detected in the bone marrow. In AA amyloidosis, elevated acute phase proteins (such as CRP and the precursor protein SAA) can be found in the blood. This is illustrative but certainly not proof of AA amyloidosis because these inflammatory proteins may also be present in the blood in many other conditions.
Although a certain type of amyloid is clinically plausible, confirmation of the amyloid type is necessary for further treatment planning. The method of treatment of the different systemic types varies widely.
Typing using the biopsy
Immunohistological examination of the biopsy offers the opportunity to further characterize the type of amyloid via specifically targeted antibodies. In AA amyloidosis, this study is sufficient for typing, provided sensitive and specific monoclonal antibodies are used. However, in AL and ATTR amyloidosis, this method fails in some cases because the antibodies used are less specific and the antigenic recognisability of the precursor protein within the amyloid deposits is reduced.
Below are examples of a renal biopsy of an AA amyloidosis patient where the biopsy is stained, on the left, with Congo Red (where the effect of the polarization filter can be seen) and on the right Immunohistochemical staining with the specific monoclonal antibody directed against Serum amyloid A (SAA ; clone Reu86.2).
 |
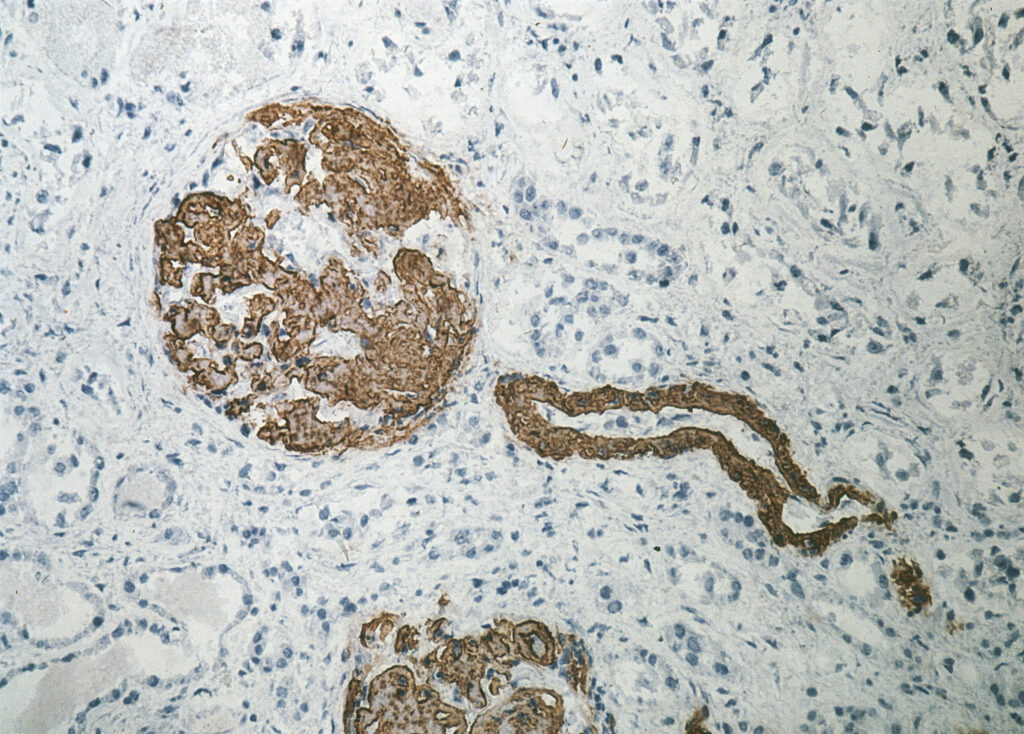 |
Kidney – Congo Red staining | Kidney – Immunohistochemical staining with anti-SAA |
In Groningen we are able to extract the amyloid from the abdominal fat biopsy and to further type the amyloid through a panel of immunochemical tests, known as ELISAs. This method is suitable for distinguishing the most common types of amyloidosis (AA, AL-kappa, AL-lambda and ATTR), but only works if there is enough amyloid in the fat biopsy. If this method fails to characterize the amyloid, other biochemical techniques such as mass spectrometry (proteomics) can be used.
Genetic testing
If there is evidence of ATTR amyloidosis, DNA testing will distinguish between the hereditary and the wild-type form. In addition to hereditary ATTR amyloidosis, there are several other, rarer types of hereditary amyloidosis (see table).
The DNA testing is done at the Department of Clinical Genetics. Most forms of hereditary amyloidosis have an autosomal dominant inheritance pattern.
When hereditary amyloidosis is diagnosed, adult family members are eligible for DNA testing. Patients with hereditary amyloidosis receive an information letter that they can give to their family members. Family members who wish to have DNA testing for hereditary amyloidosis and / or would like more information about this, can go to the Department of Clinical Genetics . If a DNA test shows that someone is a carrier of the predisposition for hereditary amyloidosis, a referral is made to the expertise center for screening for possible symptoms.
| Abbreviation | Protein (precursor) | Affected organs |
| ApoAI | Apolipoprotein A-I | Heart, liver, kidneys, nerves, larynx |
| ApoAII | Apolipoprotein A-II | Kidneys, heart |
| ApoCII | Apolipoprotein C-II | Kidneys |
| ApoCIII | Apolipoprotein C-III | Kidneys |
| Beta2M | Beta-2 microglobulin | Gastrointestinal system, autonomic nervous system, salivary and tear glands |
| CST3 | Cystatin | Brain |
| FGA | Fibrinogen Alfa chain | Kidneys |
| GSN | Gelsolin | Nervous system, skin and eyes |
| IL31RA | Interleukin-31 receptor A | Skin |
| LYS | Lysozyme | Kidneys, liver, heart, spleen, gastrointestinal tract, salivary glands |
| OSMR | Oncostatin M receptor | Skin |
| TTR | Transthyretin | Nervous system, heart, eyes, meninges |
In short
- If amyloidosis is suspected, the amyloid must be detected in a biopsy
- The type of amyloid must also be determined. Various laboratory techniques are available for this
- Genetic testing is carried out to assess whether the form is hereditary.
